
服務熱線:0755-23020893
公司座機:0755-23020893
業務直線:13530892758
公司郵箱:yjdsy88@126.com
微信平臺:億金達受控環境
公司地址:深圳市龍崗區寶荷大道76號智慧家園1棟B座1702
生產基地:東莞市常平鎮松柏塘村北橫路8號
盡管EU與FDA的GMP指南非常相似,但仍有一些地方是不同的,如年度回顧、QP制度、確認與驗證、供應商審計、交叉污染風險評估、輔料要求等。如下:
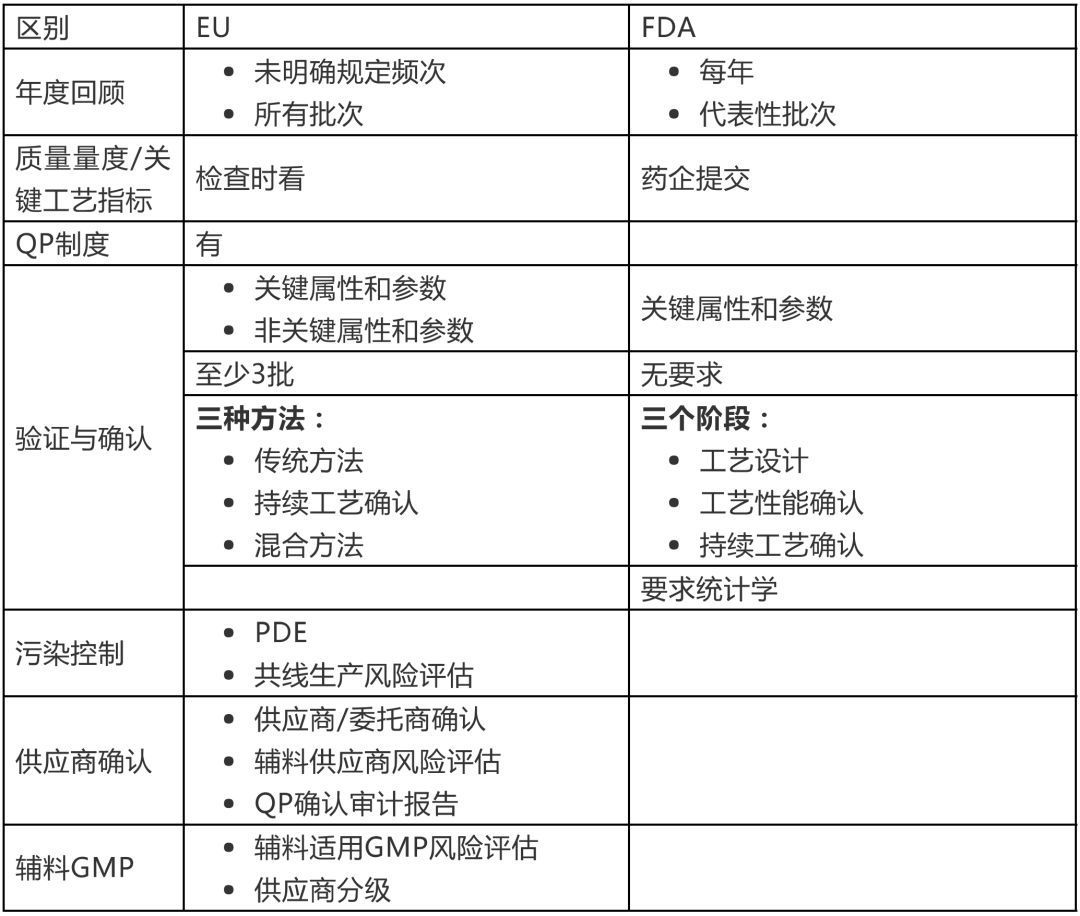
EU與FDA GMP指南的不同
QP制度
在EU,必須由指定的QP對每批(商業化或臨床試驗用)藥品的GMP符合性進行認證。QP的職責定義于EU-GMP指南附錄16中。如果商業化藥品或IMP是在美國生產或包裝,然后進口至EU,則需要在EU進行額外的檢驗。此外還需要進行供應商確認,包括有依據相應的EU-GMP指南進行的首次以及之后的定期合規審計。這些審計要由QP或其代表執行。由藥監機構執行的檢查并不能取代審計的要求。因此即使是在互認協議(MRA)全面實施之后,美國公司仍需要面對EU的審計。
在美國,質量部門負責根據CFR第211.192部分進行生產記錄審核,并確保委托生產商符合GMP要求(211.22a)。
確認與驗證
FDA工藝驗證指南與修訂后的EU GMP指南附錄15(確認與驗證)現在是越來越接近了。與FDA指南更為接近也是修訂EU-GMP指南附錄15的理由之一。
有一個差異是附錄15還要求在驗證方案中列出非關鍵屬性和參數。FDA的工藝驗證指南只要求關鍵質量屬性和關鍵工藝參數標準。在驗證批數方面也有區別。附錄15指出最少批次為3批,而FDA工藝驗證指南則并未提及批次要求。對于FDA,在工藝驗證方法上也有差異。在附錄15中提到了3種驗證方法(傳統方法、持續工藝確認和混合方法),而FDA工藝驗證指南則未作出區別。另外,對統計的要求也有區別。在FDA工藝驗證指南中對此有著更多的強調。FDA甚至建議有統計學家創建數據收集計劃,并就使用何種統計方法提供咨詢。FDA中關于工藝驗證生命周期第3階段取樣方面也有差異(持續工藝確認),要求取樣更多---至少直到有足夠的數據用于評估波動性,而在附錄15的持續工藝確認中則沒有要求增加樣品數量。
FDA對“年度產品回顧”(APR)的目的是對每種藥品的質量標準進行年度評估,并確定是否需要對質量標準或生產或控制規程進行修改。因此,需要回顧具有代表數量的批次。EU的“產品質量回顧”(PQR)則更為關注總體的生產和質量體系,并展示可持續生產出符合相應質量的產品。而且,“產品質量回顧”(PQR)要求包含所生產的所有批次。EU檢查員通常會在檢查前索取“產品質量回顧”(PQR)。
追溯&趨勢分析以及關鍵工藝指標(KPI)
根據EU-GMP第1章“適合于藥品生產的制藥質量體系應通過制定和使用有效的工藝性能和產品質量監控系統來確保建立并保持受控狀態”,使用KPI來證明受控狀態或發起和控制潛在的持續改善程序是很重要的。企業高層應定期對此進行審核(1.6)。總體來說,所有生產工藝均應“清晰定義,根據歷史數據進行系統回顧,并證實可以持續生產出所需質量并符合其標準的藥品”。
所以EU在GMP檢查過程中要看質量量度數據,而FDA則會有不同的方法。FDA于2016年起草了“行業指南:質量量度數據提交”。FDA希望在其實施之后,生產商會以電子形式向FDA提交規定的質量量度數據。FDA將以此計算特定的統計指標,進而制定基于風險的檢查計劃。
EU特有
污染控制
2015年,EU-GMP指南第3章和第5章修訂,對污染控制關注頗多。在新的第3章“設備設施”中的主要變化是防止交叉污染的相關措施。該變化是與第5章(生產)的修訂以及EMA關于多產品共用設施設定基于健康暴露限風險識別指南(EMA/CHMP/CVMP/SWP/169430/2012)緊密相關的,新的內容要求以毒性數據為基礎基于風險進行評估。這意味著通過技術或組織措施無法控制所識別的風險時,則需要使用專用設施。
這也受到上述EMA毒性指南的支持。該指南自2015年6月1日起生效,其中描述了基于共用設施/設備區域中所生產藥品的毒性評估的風險評估。修訂后的第5章“生產”亦高度關注防止交叉污染的技術和組織措施。
總的來說,需要有文件規定污染控制策略,并應使用QRM原則評估和控制污染&交叉污染的風險。
供應鏈追溯
對(所有)供應商進行確認對于上市許可持有人MAH來說是法定義務。在申報MA時,生產商就需要對相應的API供應商進行審計(和確認)。并且這只是持續確認的開始。
EU指令2001/83/EC第46條要求藥品生產商應通過對生產和分銷設施進行審計以確認其所用API生產商的合規性(以符合GMP指南)。對于輔料供應商,至少應有正式的風險評估(參見下一段)。
EU-GMP指南第7章(外包活動)要求在委托活動之前“委托方有義務評估受托方成功執行外包活動的合法性、適當性及其資質”【7.5】,并且“委托方應監管和審核受托方的表現(……)”【7.7】。
根據EU-GMP指南附錄16的要求,QP必須確保“已審計了藥品生產和檢驗相關的所有場所,以及API生產場所,并且執行認證的QP應可以獲得這此審計報告”【1.7.3】。
輔料GMP
歐盟2015年3月19日發布了一份藥用輔料供應商的GMP指南“,確定人用藥輔料適用GMP的正式風險評估指南”。要求是生產許可持有人進行風險評估以評價與輔料生產商/供應商有關的所有風險,以確定適當的GMP控制措施并對生產商的風險概況進行分級用于供應商確認。
